藥品多產(chǎn)品共線生產(chǎn)的質(zhì)量與安全管控是制藥行業(yè)面臨的重要課題��。每日允許暴露量(PDE)評估作為核心技術(shù)支撐�����,對共線生產(chǎn)風(fēng)險識別與控制具有關(guān)鍵意義。本文基于風(fēng)險管理理念�����,系統(tǒng)構(gòu)建了藥品共線生產(chǎn)風(fēng)險評估體系:首先明確了 PDE 的獲取方式及多文獻(xiàn)數(shù)據(jù)差異的優(yōu)先級判斷原則����,在此基礎(chǔ)上從法規(guī)符合性�、產(chǎn)品預(yù)定用途、產(chǎn)品特性��、共線產(chǎn)品廠房設(shè)施設(shè)備四個核心維度開展全面評估����。其中,法規(guī)符合性評估依據(jù) GMP 及相關(guān)指南明確了禁限共線品類及不同 PDE 值產(chǎn)品的生產(chǎn)要求�;產(chǎn)品預(yù)定用途評估聚焦于藥物相互作用及氧化還原反應(yīng)風(fēng)險;產(chǎn)品特性評估重點分析物料特性����、活性 /毒性及殘留限度計算;廠房設(shè)施設(shè)備分析則從多風(fēng)險點識別出發(fā)確定共線可行性��。
結(jié)合小容量注射劑生產(chǎn)實踐�,進(jìn)一步制定了涵蓋生產(chǎn)線管控、清潔驗證 / 確認(rèn)、品種動態(tài)管理���、多區(qū)域污染控制及 OEB/PDE 層面的全鏈條控制策略���。研究表明,該評估體系與控制策略可有效降低共線生產(chǎn)的污染���、交叉污染及混淆風(fēng)險�,保障藥品質(zhì)量安全���。但受產(chǎn)品特性復(fù)雜性影響�,風(fēng)險評估在量化預(yù)測方面仍存在局限性����,未來需通過積累更多生產(chǎn)數(shù)據(jù)、深入研究產(chǎn)品特性影響機(jī)制以提升評估精準(zhǔn)性���。
Part0前言
每日允許暴露量(英文 Permitted Daily Exposure����,簡稱 PDE)是指在人的終生時長內(nèi)����,藥物或產(chǎn)品每日可接受的最大攝入量�。對于制藥企業(yè)而言��,在生產(chǎn)線引入新產(chǎn)品時�����,均需計算����、審核和評估藥品 PDE�����,一是確認(rèn)該產(chǎn)品 PDE 是否科學(xué)�、權(quán)威、專業(yè)�,二是判斷引入該產(chǎn)品是否能與現(xiàn)有生產(chǎn)線產(chǎn)品共線生產(chǎn),三是用于制定設(shè)備清潔殘留限度��、雜質(zhì)限度����、生產(chǎn)環(huán)境殘留物限度的可接受標(biāo)準(zhǔn)�。
制藥企業(yè)多數(shù)會采用委托第三方權(quán)威機(jī)構(gòu)或毒理學(xué)專家計算����,或通過權(quán)威監(jiān)管機(jī)構(gòu)文獻(xiàn)等方式獲取 PDE。在實際毒理評估工作中�,如果同一物質(zhì)在多份文獻(xiàn)中出現(xiàn)且數(shù)值不一致或存在差異,通常會按照文獻(xiàn)的權(quán)威性�����、數(shù)據(jù)原始程度��、GLP 合規(guī)程度來進(jìn)行優(yōu)先級判斷��。
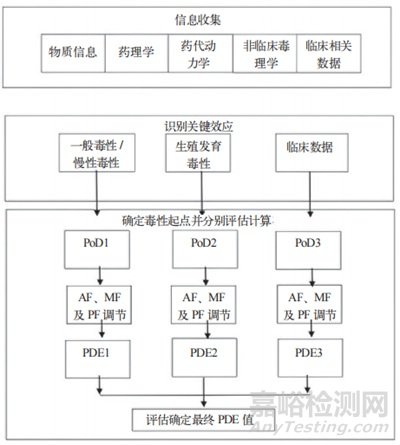
開展藥品每日允許暴露量(PDE)評估時���,首先應(yīng)充分收集藥品活性成分的非臨床試驗數(shù)據(jù)和人體臨床使用數(shù)據(jù)����。接著�,通過數(shù)據(jù)評估識別關(guān)鍵效應(yīng),并選出相應(yīng)的毒性起始點(PoD)��。然后�,根據(jù)相關(guān)指導(dǎo)原則和本標(biāo)準(zhǔn)的評估原則設(shè)置調(diào)節(jié)因子及其他因子����,并依據(jù)公式計算得到 PDE 數(shù)值����。這一流程確保了 PDE 評估的科學(xué)性和準(zhǔn)確性,為藥品的生產(chǎn)����、研究和檢測活動提供風(fēng)險評估和管理的依據(jù)。
Part1基于風(fēng)險管理的藥品共線生產(chǎn)風(fēng)險評估
啟動的共線評估報告 [1] 應(yīng)包含法規(guī)符合性�、產(chǎn)品工藝、產(chǎn)品預(yù)定用途���、產(chǎn)品基本信息以及殘留限度(含溶解度最差、PDE 最小���、殘留限度最?��。⒐簿€產(chǎn)品廠房 / 設(shè)備 / 設(shè)施共用等多個方面��。首先需要收集共線評估信息���,包含的內(nèi)容有:劑型�、產(chǎn)品規(guī)格、最小 / 最大批量��、給藥途徑���、臨床適應(yīng)癥��、藥理作用����、用藥禁忌�����、配伍禁忌����、用藥對象、用藥劑量����、長期用藥(藥品在體內(nèi)是否蓄積并產(chǎn)生毒性)、產(chǎn)品類別����、活性成分通用名或化學(xué)名�、致敏性�、活性微生物、原輔料性狀(包含顏色����、氣味、狀態(tài)等)�、原輔料溶解度、毒理相關(guān)數(shù)據(jù)(毒性試驗數(shù)據(jù)��、ED50��、LD50�、PDE/ ADE、OEL 等)��、清潔劑等���。共線評估的風(fēng)險評估方法采用 ICH Q9 的工具進(jìn)行,本文不進(jìn)行闡述���。
根據(jù)《藥品共線生產(chǎn)質(zhì)量風(fēng)險管理指南》[2] 規(guī)定�����,PDE ≤ 10 μg/ 天(具有細(xì)胞毒性類)的化學(xué)藥品目前只能在高毒性藥品專用生產(chǎn)車間生產(chǎn)�����;PDE ≤ 10 μg/ 天(無細(xì)胞毒性)的產(chǎn)品不能在普通化藥車間共線生產(chǎn)�����,生產(chǎn)時需用專用設(shè)備(如采取一次性配液系統(tǒng))���;PDE在 10 ~ 100 μg/ 天(包含 100 μg/ 天)范圍內(nèi)�,若為在研產(chǎn)品的試制批����、臨床試驗用藥品,應(yīng)執(zhí)行清潔殘留分析方法的轉(zhuǎn)移確認(rèn)或驗證�����、清潔確認(rèn)�;若為在研產(chǎn)品的驗證批,應(yīng)執(zhí)行清潔殘留分析方法的轉(zhuǎn)移確認(rèn)或驗證、清潔驗證���;若為商業(yè)批�,按照已有的清潔驗證參數(shù)執(zhí)行清洗����,并進(jìn)行定期清潔再驗證。除以上措施外���,還會采取階段性生產(chǎn)方式���,若有必要可采取一次性配液系統(tǒng)或?qū)χ苯咏佑|藥液的器具進(jìn)行專用化、限定化排產(chǎn)等�����。
PDE 報告至少包含以下內(nèi)容:(1)物質(zhì)一般信息:化學(xué)性質(zhì)��、化合物名稱��、CAS 號��、相關(guān)理論數(shù)據(jù)等�;(2)化合物的藥理作用機(jī)制(MOA)及適應(yīng)癥����;(3)非臨床數(shù)據(jù):藥理學(xué)和安全藥理學(xué)數(shù)據(jù)���、藥物動力學(xué)和代謝、毒理數(shù)據(jù)����、單劑量毒性、重復(fù)給藥毒性���、基因毒性�、致癌性�、發(fā)育和生殖毒性、局部耐受 /(皮膚)致敏����;(4)人類的數(shù)據(jù):人的治療劑量(所有適應(yīng)癥、途徑和患者人群�,包括使用的易感人群、最低藥理活性劑量)和 / 或臨床試驗數(shù)據(jù)�、藥物代動力學(xué)和藥物 - 藥物相互作用、臨床試驗 / 治療使用中的不良反應(yīng)���,包括禁忌癥和注意事項�����、妊娠和哺乳期資料���;(5)PDE/ADE 詳細(xì)計算過程�;(6)PDE/ADE 詳細(xì)選擇過程和結(jié)論�����;(7)參考文獻(xiàn)和參與評估的專家列表�。
開展藥品每日允許暴露量(PDE)評估時,首先需充分收集藥品活性成分的非臨床試驗數(shù)據(jù)和人體臨床使用數(shù)據(jù)����。接著,通過數(shù)據(jù)評估識別關(guān)鍵效應(yīng)����,并選出相應(yīng)的毒性起始點(PoD)。然后�,根據(jù)相關(guān)指導(dǎo)原則和本標(biāo)準(zhǔn)的評估原則設(shè)置調(diào)節(jié)因子及其他因子,并依據(jù)公式計算得到 PDE 數(shù)值���。這一流程確保了 PDE 評估的科學(xué)性和準(zhǔn)確性����,為藥品的生產(chǎn)�����、研究和檢測活動提供風(fēng)險評估和管理的依據(jù)�,圖 1 所示為 PDE 評估的一般流程。
圖 1 PDE 評估的一般流程 [3]
Part2PDE 在產(chǎn)品特性中的評估
產(chǎn)品特性評估是通過比對擬共線生產(chǎn)的多個產(chǎn)品的各項特性����,以評估是否具備共線生產(chǎn)的可能性。擬共線生產(chǎn)品種特性相似或相近����,可多產(chǎn)品共線;若擬共線生產(chǎn)品種特性具有特殊性�����,則不可多產(chǎn)品共線���。產(chǎn)品特性評估主要包括以下內(nèi)容���。
(1)將產(chǎn)品類別��、生產(chǎn)過程中所用物料的特性 [4](包括物料狀態(tài)如粉末�����、液態(tài)����、固態(tài)�����、性狀如顏色����、氣味、黏度����,溶解度等具體數(shù)據(jù))輸出至車間原輔料溶解度信息表,對比各產(chǎn)線上品種的物料差異性���。存在特殊性質(zhì)(如溶解度較差不易清洗)的品種��,應(yīng)執(zhí)行清潔殘留分析方法的轉(zhuǎn)移確認(rèn)或驗證����、清潔確認(rèn)或清潔驗證等風(fēng)險控制措施。
(2)對產(chǎn)品的活性 / 毒性(如是否具有細(xì)胞毒性���、致癌性,PDE/ADE�����、OEL���、TTC 等)����、致敏性����、活性微生物(疫苗或病毒)進(jìn)行評估。依據(jù)《藥品共線生產(chǎn)質(zhì)量風(fēng)險管理指南》的要求����,化學(xué)物質(zhì)基于健康的暴露限度(HBEL)在評估清潔殘留數(shù)據(jù)時更具科學(xué)性和優(yōu)勢�。在評價 HBEL 時��,每日允許暴露量(Permitted Daily Exposure���,PDE)及每日可接受暴露量(Acceptable Daily Exposure�,ADE)是被普遍接受的標(biāo)準(zhǔn)���。將產(chǎn)品的 PDE(若無 PDE�,需提供藥理毒理資料)及最大日劑量等數(shù)據(jù)輸出至車間產(chǎn)品基本信息表�����。若無 PDE����,則需根據(jù)最嚴(yán)格的毒理學(xué)關(guān)注閾值 TTC 值 1.5 μg/ 天進(jìn)行計算,該標(biāo)準(zhǔn)源自 ICH M7(R1):評估和控制藥物中DNA 反應(yīng)性(致突變)雜質(zhì)以限制潛在致癌風(fēng)險�����。隨后�����,對比各產(chǎn)線上的 PDE 最小品種,對產(chǎn)線中 PDE 最小的品種執(zhí)行清潔殘留分析方法的轉(zhuǎn)移確認(rèn)或驗證�����、清潔確認(rèn)或清潔驗證等風(fēng)險控制措施����。
(3)引入品種采用最科學(xué)的方法(PDE 法)計算最小允許殘留總量,將共線評估需求表中已有信息(如產(chǎn)品最大日劑量����、批量��、理論濃度等數(shù)據(jù))輸入至車間計算矩陣表�,并由另一人復(fù)核輸入數(shù)據(jù)的正確性。同時使用傳統(tǒng)方法“1/1000 最低日劑量法����、10PPM”計算最小允許殘留總量。若傳統(tǒng)方法計算的殘留總量低于 PDE法�,如有必要,可將傳統(tǒng)方法計算的殘留限度作為警戒限��;若傳統(tǒng)方法計算的殘留總量高于 PDE 法�,則繼續(xù)沿用 PDE 法作為最小允許殘留總量���。
Part3清潔驗證 / 確認(rèn)中 PDE 的應(yīng)用控制策略
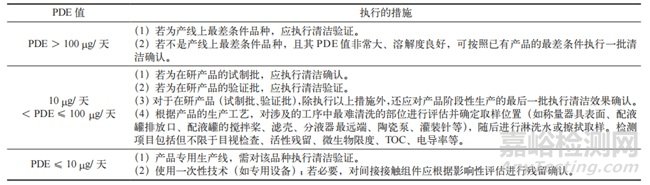
基于 PDE 值的清潔驗證控制策略的核心在于根據(jù)化合物的毒理學(xué)數(shù)據(jù)、產(chǎn)品類型及生產(chǎn)階段���,實施分級管理與動態(tài)調(diào)整���,具體應(yīng)用體現(xiàn)在以下幾個方面。表 1所示為不同 PDE 值產(chǎn)品的清潔驗證控制策略舉例�。
表 1 不同 PDE 值產(chǎn)品的清潔驗證控制策略舉例
(1)對于上市產(chǎn)品,應(yīng)根據(jù)產(chǎn)品的年度質(zhì)量回顧及評估制訂清潔再驗證周期 [5]�。對于 10 μg/ 天<PDE ≤ 100 μg/ 天的品種,應(yīng)在階段性生產(chǎn)的最后一批執(zhí)行清潔效果確認(rèn)����;對該產(chǎn)線上市產(chǎn)品中條件最差的品種,每半年執(zhí)行一次清潔效果確認(rèn)���。根據(jù)產(chǎn)品的生產(chǎn)工藝�����,應(yīng)對涉及的工序中最難清洗的部位進(jìn)行評估并確定取樣位置(如稱量器具表面�、配液罐排放口、配液罐的攪拌槳��、濾殼�����、分液器最遠(yuǎn)端��、陶瓷泵�����、灌裝針等)進(jìn)行淋洗水或擦拭取樣����,檢測項目包括但不限于目視檢查���、活性殘留����、微生物限度��、總有機(jī)碳(TOC)���、電導(dǎo)率等�����。
(2)對于在研產(chǎn)品(試制批����、驗證批),均需執(zhí)行清潔確認(rèn)或清潔驗證品種�����。此外���,還需執(zhí)行清潔殘留分析方法的轉(zhuǎn)移確認(rèn)或驗證����。對于同品種�����、同工藝的產(chǎn)品��,若在產(chǎn)線上已有品種進(jìn)行清潔驗證�����,則可視為擬引入的品種已執(zhí)行清潔驗證,但后續(xù)轉(zhuǎn)商業(yè)化生產(chǎn)時需重新評估����。
(3)對于臨床試驗用藥品,每批次均需執(zhí)行清潔確認(rèn)����,后續(xù)轉(zhuǎn)商業(yè)化生產(chǎn)時需重新評估。
(4)首次進(jìn)行清潔驗證或確認(rèn)時�����,需根據(jù)產(chǎn)品的生產(chǎn)工藝�,對涉及的工序中最難清洗的部位進(jìn)行評估并確定取樣位置進(jìn)行淋洗水或擦拭取樣;在進(jìn)行再驗證或再確認(rèn)�、階段性生產(chǎn)清潔確認(rèn)時,可根據(jù)首次清潔驗證或確認(rèn)的情況對最難清洗部位進(jìn)行取樣評估����。取樣檢測項目包括但不限于活性殘留等項目��。
Part4基于 OEB/PDE 層面建立控制策略舉例
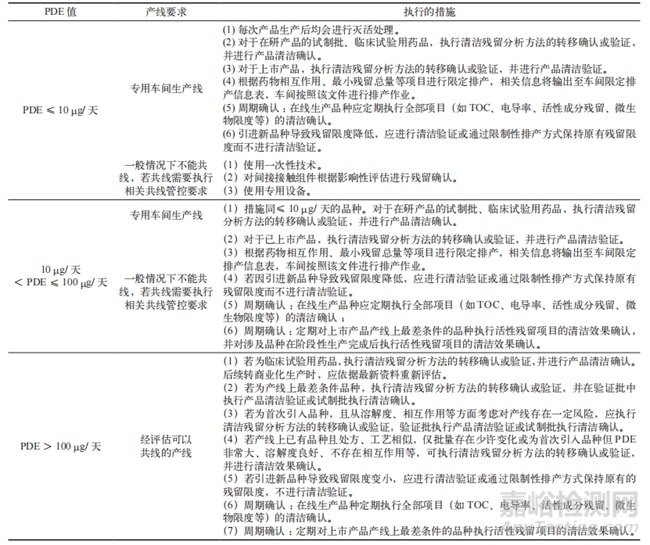
為將基于 OEB/PDE 的風(fēng)險分級原則轉(zhuǎn)化為具體�、可執(zhí)行的控制方案,可依據(jù)不同的 PDE 范圍,對生產(chǎn)線的使用策略及清潔驗證等控制措施進(jìn)行差異化設(shè)計����,具體示例如表 2 所示。
表 2 基于 OEB/PDE 層面建立控制策略舉例
Part5結(jié)論與展望
風(fēng)險評估和控制策略是藥品共線生產(chǎn)在小容量注射劑中實踐的關(guān)鍵 [6]�����。在風(fēng)險評估方面����,通過準(zhǔn)確計算藥品每日允許暴露量(PDE),全面評估法規(guī)符合性��、產(chǎn)品預(yù)定用途����、產(chǎn)品特性以及共線產(chǎn)品廠房設(shè)施設(shè)備等因素,可以有效識別共線生產(chǎn)中的風(fēng)險點����。在控制策略方面,制定科學(xué)合理的生產(chǎn)線控制策略�����、清潔驗證 / 確認(rèn)的控制策略、生產(chǎn)線品種動態(tài)管理策略�����,以及從生產(chǎn)區(qū)�、倉儲區(qū)、檢驗區(qū)層面和基于 OEB/PDE 層面建立控制策略��,能夠有效降低風(fēng)險����,保障藥品質(zhì)量和生產(chǎn)安全。
在風(fēng)險評估的精準(zhǔn)性上��,雖然采用了多種評估方法和工具��,但由于共線生產(chǎn)涉及的因素復(fù)雜多樣�����,不同產(chǎn)品的特性差異顯著��,如活性成分的結(jié)構(gòu)���、穩(wěn)定性和代謝途徑各不相同��,使得風(fēng)險評估在量化和預(yù)測風(fēng)險方面仍存在一定的局限性����。應(yīng)進(jìn)一步深入研究不同產(chǎn)品特性對共線生產(chǎn)風(fēng)險的影響機(jī)制�,可通過收集大量的藥品共線生產(chǎn)數(shù)據(jù),包括產(chǎn)品特性���、生產(chǎn)工藝參數(shù)����、設(shè)備運(yùn)行數(shù)據(jù)等���,實現(xiàn)風(fēng)險的準(zhǔn)確預(yù)測和評估�����。
參考文獻(xiàn)
[1] 田文淼���,梁毅. 小容量注射劑共線生產(chǎn)的質(zhì)量風(fēng)險控制研究[J]. 中國醫(yī)藥工業(yè)雜志,2020�,51(6):789-794.DOI:10.16522/j.cnki.cjph.2020.06.019.
[2] 國家藥品監(jiān)督管理局食品藥品審核查驗中心. 藥品共線生產(chǎn)質(zhì)量風(fēng)險管理指南[EB/OL] (2023-03-06) [2025-5-12].
[3] 浙江省藥品監(jiān)督管理與產(chǎn)業(yè)發(fā)展研究會. 藥品每日允許暴露量評估方法:標(biāo)準(zhǔn)號T/ZJDAIR 011-2024[S/OL].(2025-04-28)[2025-05-12].
[4] 國家藥典委員會. 中華人民共和國藥典:2025年版·四部[M]. 北京:中國醫(yī)藥科技出版社,2025:3-4.
[5] 廣東省藥品監(jiān)督管理局. 關(guān)于進(jìn)一步加強(qiáng)廣東省藥品委托生產(chǎn)監(jiān)督管理有關(guān)事項的通知[EB/OL]. (2024-07-12) [2025-5-12].
[6] 中華人民共和國衛(wèi)生部. 藥品生產(chǎn)質(zhì)量管理規(guī)范(2010年修訂):衛(wèi)生部令第79號[EB/OL].(2011-01-17 )[2025-5-12].