為何不將口服藥物進(jìn)入體內(nèi)后的旅程��,比作一場驚心動(dòng)魄的��、充滿未知與挑戰(zhàn)的“歷險(xiǎn)記”呢��?或許這樣可以更加有利于讓我們理解藥物的吸收過程以及在藥物開發(fā)中應(yīng)該關(guān)注哪些重要的方面��,為更好的為藥物吸收奠定基礎(chǔ)��。一枚小小的藥片,從被患者吞下的那一刻起��,便踏上了一條通往血液循環(huán)系統(tǒng)的崎嶇之路��。這條路上��,它要經(jīng)受胃酸的洗禮��,穿越復(fù)雜的黏液層,突破由緊密連接的細(xì)胞構(gòu)成的腸道屏障��,還要設(shè)法逃脫無處不在的代謝酶的“抓捕”和外排轉(zhuǎn)運(yùn)蛋白的“驅(qū)逐”��。最終��,即使僥幸進(jìn)入門靜脈,它還將面臨肝臟這一終極代謝器官的“首過效應(yīng)”考驗(yàn)��。這場旅程的成敗��,直接決定了藥物的口服生物利用度��,也是許多藥物開發(fā)項(xiàng)目最終折戟沉沙的主要原因��。據(jù)統(tǒng)計(jì)��,在新藥開發(fā)失敗案例中��,40%~50%的藥物研發(fā)臨床失敗是由于缺乏療效��,這直接與口服生物利用度不足相關(guān),這使其成為制約候選藥物成功轉(zhuǎn)化為上市產(chǎn)品的關(guān)鍵瓶頸��。上世紀(jì)90年代��,有30~40%的藥物研發(fā)失敗是由于所選的先導(dǎo)化合物類藥性不佳��。而目前失敗率已經(jīng)降至10%左右��,正是得益于類藥性的選擇��,包括溶解度��、滲透性、蛋白質(zhì)結(jié)合��、代謝穩(wěn)定性和體內(nèi)藥代動(dòng)力學(xué)如生物利用度等��。
<span lang="EN-US" style="font-family:"Times New Roman",serif; mso-fareast-font-family: <p>宋體; mso-no-proof:yes; "><span leaf=;" "="">
圖1 藥物吸收過程
歷險(xiǎn)的起點(diǎn)始于胃部��。當(dāng)患者口服藥物后��,固體制劑首先進(jìn)入這個(gè)強(qiáng)酸性的環(huán)境(pH1.2-2.0)。對(duì)于許多藥物而言��,這里不啻為第一道“鬼門關(guān)”��。酸敏感藥物可能在此發(fā)生顯著降解��,失去活性。制劑本身也在此開始了它的“變形記”��,經(jīng)歷崩解和解聚過程,從完整的片劑或膠囊分散為更小的顆粒��,這一步驟至關(guān)重要��,因?yàn)樗鼧O大地增加了藥物與胃液的接觸面積,為后續(xù)的溶解過程奠定基礎(chǔ)��。然而��,胃部強(qiáng)大的排空能力意味著留給藥物停留的時(shí)間非常有限��,且其黏膜層較厚��,并非吸收的主要場所��。
圖2 腸道菌群–藥物相互作用及其對(duì)藥物生物利用度影響的示意圖。主要細(xì)菌門類(厚壁菌門��、擬桿菌門和放線菌門)通過直接機(jī)制參與藥物代謝,包括還原反應(yīng)��、水解反應(yīng)和去結(jié)合反應(yīng)。飲食��、年齡、疾病狀態(tài)��、抗生素暴露及宿主遺傳等多種因素調(diào)節(jié)著這些菌群–藥物的相互作用。這些相互作用的共同影響最終體現(xiàn)為對(duì)藥物關(guān)鍵性質(zhì)的調(diào)控��,包括溶解度��、吸收效率及整體生物利用度。
真正的考驗(yàn)與機(jī)遇在小腸��。當(dāng)藥物隨胃排空進(jìn)入十二指腸,便來到了人體為吸收而設(shè)計(jì)的“主戰(zhàn)場”��。小腸擁有巨大的表面積(因絨毛和微絨毛結(jié)構(gòu))��、更適宜且呈梯度變化的pH環(huán)境(從十二指腸的pH4-5到回腸末端的pH8)以及豐富的血流灌注��。在這里��,藥物分子必須完成從固態(tài)到溶液態(tài)的轉(zhuǎn)變��,即“溶解”��,因?yàn)橹挥腥芙夂蟮姆肿討B(tài)藥物,才具備穿越細(xì)胞膜的基本資格��。對(duì)于難溶性藥物(BCS II類和IV類),這一步本身就是巨大的挑戰(zhàn)。它們的溶解速率和溶解度極低��,導(dǎo)致吸收窗口期內(nèi)能溶解的藥物量不足��,大部分有效成分只能隨著腸道蠕動(dòng)被排出體外��,這是導(dǎo)致生物利用度低下的核心原因之一��。
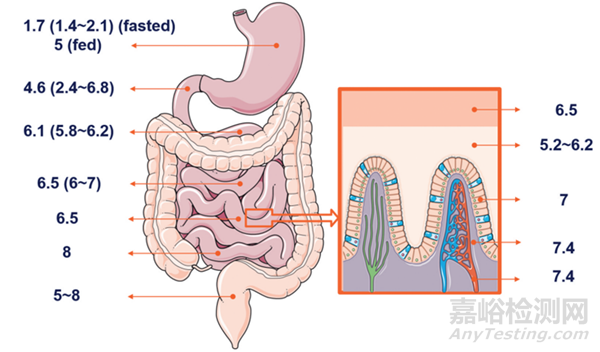
圖3 胃腸道pH
成功溶解的藥物分子��,此刻正面臨那道最關(guān)鍵��、最復(fù)雜的物理屏障——腸道上皮細(xì)胞層��。這是一道由單層細(xì)胞構(gòu)成的“城墻”,細(xì)胞之間被名為“緊密連接”的蛋白質(zhì)復(fù)合體牢牢封死��,僅允許極小的水溶性分子通過��。藥物要想進(jìn)入血液循環(huán),主要有兩條路徑��。其一是“跨細(xì)胞途徑”��,即直接穿透細(xì)胞膜的脂質(zhì)雙分子層��,這主要依賴于藥物的脂溶性和解離狀態(tài)��。根據(jù)pH分配假說��,弱酸性藥物在酸性胃液中更易以非離子型存在,利于跨膜��,但小腸巨大的表面積使其仍成為大多數(shù)藥物(無論酸堿性)吸收的主要場所��。其二是“旁細(xì)胞途徑”��,即從細(xì)胞間的緊密連接縫隙中鉆過去,但這條路徑對(duì)分子大小和電荷要求極為苛刻��,并非主流��。
就在藥物即將穿越細(xì)胞時(shí),它可能遭遇另一類關(guān)鍵角色的攔截——外排轉(zhuǎn)運(yùn)蛋白��,其中最具代表性的便是P-糖蛋白(P-gp)和乳腺癌耐藥蛋白(BCRP)��。這些蛋白如同“看門人”或“清道夫”,它們位于腸上皮細(xì)胞的頂端膜上��,一旦識(shí)別出它們“眼中”的外來物(即藥物的底物)��,便會(huì)利用ATP水解產(chǎn)生的能量,主動(dòng)將藥物從細(xì)胞內(nèi)泵回腸腔��,從而極大地限制藥物的吸收��。許多藥物��,如地高辛��、環(huán)孢素以及部分抗癌藥,都是P-gp的底物��。研究表明��,當(dāng)同時(shí)使用P-gp抑制劑(如維拉帕米)時(shí)��,其底物藥物的口服生物利用度會(huì)顯著提高��,這清晰地揭示了這些“看門人”在吸收過程中的決定性作用��。
即便藥物幸運(yùn)地躲過了外排轉(zhuǎn)運(yùn)蛋白的“驅(qū)逐”��,成功進(jìn)入了腸上皮細(xì)胞內(nèi)部,它依然身處險(xiǎn)境��。細(xì)胞內(nèi)存在著各種藥物代謝酶��,尤其是細(xì)胞色素P450酶系(如CYP3A4)��,它們可以對(duì)藥物進(jìn)行結(jié)構(gòu)修飾,即發(fā)生“腸代謝”��,使藥物在尚未進(jìn)入血液循環(huán)前就被滅活��。這是藥物吸收后遭遇的“第一重代謝”考驗(yàn)��。只有那些經(jīng)受住這一切,最終從細(xì)胞基底側(cè)膜成功“出逃”的藥物分子��,才能進(jìn)入細(xì)胞間隙��,并最終被吸收進(jìn)入?yún)R入門靜脈的毛細(xì)血管��。
然而��,歷險(xiǎn)記并未就此結(jié)束。門靜脈血液將攜帶藥物直接輸送至肝臟��。在這里��,藥物將面臨最為嚴(yán)峻的終極考驗(yàn)——肝臟首過效應(yīng)��。肝臟是人體最大的代謝器官��,富含種類繁多��、活性強(qiáng)大的藥物代謝酶��。藥物在此處經(jīng)歷一場大規(guī)模的生物轉(zhuǎn)化,或被徹底滅活,或被轉(zhuǎn)化為易于排泄的水溶性代謝產(chǎn)物��。只有逃脫了肝臟首過代謝“洗劫”的那部分原形藥物��,才能隨肝靜脈進(jìn)入下腔靜脈��,最終抵達(dá)全身血液循環(huán)��,真正完成其體內(nèi)吸收的歷險(xiǎn),奔赴靶點(diǎn)發(fā)揮作用��。
這場歷險(xiǎn)的復(fù)雜性遠(yuǎn)不止于此��。胃腸道環(huán)境本身就是一個(gè)動(dòng)態(tài)變化的生態(tài)系統(tǒng)��。食物的攝入可以顯著改變胃排空速率��、腸道pH值��,甚至通過促進(jìn)膽汁分泌形成膠束來增溶脂溶性藥物(如灰黃霉素)��。腸道血流灌注的多少��,也直接影響著藥物被吸收的速率��。此外��,數(shù)以萬億計(jì)的腸道微生物構(gòu)成了另一道生物屏障��,它們自身所攜帶的酶系也可能代謝藥物,進(jìn)一步增加了吸收過程的不確定性��。
表1人體胃腸道的生理特征
面對(duì)如此充滿艱險(xiǎn)的旅程��,藥物科學(xué)家們并未止步��,而是開發(fā)出各種精巧的制劑策略��,旨在幫助藥物“武裝”自己��,順利完成這場歷險(xiǎn)��。對(duì)于難溶性的BCS II類藥物��,科學(xué)家們通過制備納米晶體來極大增加比表面積��,提高溶解速率��;或者采用固體分散體技術(shù)��,將藥物以高能無定形態(tài)分散于親水性載體中,使其溶出就像“方糖溶于水”一樣迅速��。而對(duì)于滲透性差的BCS III類和IV類藥物��,這場“破壁之戰(zhàn)”則更為關(guān)鍵。最具里程碑意義的突破當(dāng)屬口服司美格魯肽片(Rybelsus®)的成功��。它采用了名為SNAC的吸收促進(jìn)劑��。SNAC在胃內(nèi)局部升高pH��,保護(hù)多肽藥物不被降解��,同時(shí)能增加細(xì)胞膜流動(dòng)性,促進(jìn)藥物以跨細(xì)胞途徑被吸收��,從而讓曾經(jīng)只能注射的大分子多肽藥物實(shí)現(xiàn)了口服給藥��。另一個(gè)成功的例子是口服奧曲肽(Mycapssa®)��,它利用中鏈脂肪酸辛酸鈉可逆地打開小腸上皮細(xì)胞間的緊密連接��,為藥物開辟了一條臨時(shí)的“旁細(xì)胞通道”��。還有脂質(zhì)遞送系統(tǒng)��,如自乳化給藥系統(tǒng)��,不僅能增溶,其組分本身也具有一定的促滲作用��,甚至能引導(dǎo)藥物進(jìn)入淋巴系統(tǒng)��,巧妙地繞過肝臟的首過代謝��。此外��,前藥策略則是在分子層面進(jìn)行修飾��,通過連接特定的轉(zhuǎn)運(yùn)體底物��,利用腸道上皮細(xì)胞上的攝取轉(zhuǎn)運(yùn)蛋白(如PepT1)主動(dòng)將藥物“請(qǐng)”進(jìn)細(xì)胞內(nèi)��,變被動(dòng)為主動(dòng)。
還必須指出的是��,這場歷險(xiǎn)的劇本在不同人群中并非千篇一律。對(duì)于老年患者��,胃酸分泌減少可能導(dǎo)致胃內(nèi)pH升高��,影響弱酸性藥物的溶解��;胃排空和腸道蠕動(dòng)減緩,可能延長藥物滯留時(shí)間��,也增加了酸降解風(fēng)險(xiǎn)��;腸道血流減少則可能減慢吸收速率��。對(duì)于肝功能不全的患者��,肝臟的首過代謝能力下降��,某些藥物的生物利用度反而可能異常升高,帶來中毒風(fēng)險(xiǎn)��。腎功能不全的患者��,不僅排泄受阻��,尿毒癥毒素蓄積還會(huì)損傷腸道屏障功能��,改變轉(zhuǎn)運(yùn)蛋白的表達(dá)��,從而影響藥物的吸收過程��。這些特殊人群的生理改變,使得藥物的體內(nèi)歷險(xiǎn)充滿了更多變數(shù)��,也對(duì)個(gè)體化用藥提出了更高要求。
總而言之��,一枚小小的口服藥片從入口到發(fā)揮療效,其體內(nèi)吸收歷程是一場需要跨越重重生理屏障��、歷經(jīng)多重生化考驗(yàn)的復(fù)雜冒險(xiǎn)��。對(duì)這場歷險(xiǎn)中每一個(gè)環(huán)節(jié)——從制劑的崩解、藥物的溶解��,到跨膜轉(zhuǎn)運(yùn)的機(jī)制��,再到代謝酶和外排泵的攔截——的深刻理解��,正是我們藥物研發(fā)科學(xué)家不斷優(yōu)化分子結(jié)構(gòu)��、設(shè)計(jì)精巧制劑��、最終將“歷險(xiǎn)”導(dǎo)向成功終點(diǎn)的智慧源泉��。未來��,隨著我們對(duì)腸道微觀環(huán)境、轉(zhuǎn)運(yùn)蛋白和代謝酶網(wǎng)絡(luò)的認(rèn)知日益精進(jìn)��,結(jié)合PBPK模型等先進(jìn)工具的應(yīng)用��,我們將能為每一位患者��、每一類特殊人群��,繪制出更加精準(zhǔn)��、安全的藥物體內(nèi)歷險(xiǎn)航圖��,將更多“不可能口服”的藥物��,變成患者手中便捷有效的治療武器。
胃腸道之復(fù)雜導(dǎo)致我們看到的不同來源的研究結(jié)果可能略有出處��,正如此文所展現(xiàn)的��,如果與您這邊的認(rèn)識(shí)有所不同��,請(qǐng)以個(gè)人認(rèn)知為準(zhǔn)��!如果覺得文章卻又些許啟示,還請(qǐng)點(diǎn)贊轉(zhuǎn)發(fā)��,感謝!
參考文獻(xiàn)
1.Overcoming Challenges in Small-Molecule Drug Bioavailability: A Review of Key Factors and Approaches
2. Why 90% of clinical drug development fails and how to improve it?
3. Crosstalk of physiological pH and chemical pKa under the umbrella of physiologically based pharmacokineticmodeling of drug absorption, distribution, metabolism,excretion, and toxicity
4.《默沙東診療手冊》
5. Advances in Oral Drug Delivery